秦皇岛一类医疗器械备案办理费用,一条龙服务
价格:面议 2023-08-09 05:39:01 299次浏览临床评价资料指的是? 回答: 临床评价资料需包含以下内容: ①详述产品预期用途,包括产品所提供的功能,并可描述其适用的医疗阶段(如后的监测、康复等),目标用户及其操作该产品应具备的技能/知识/培训;预期与其组合使用的器械。 ②详述产品预期使用环境,包括该产品预期使用的地点如医院、医疗/临床实验室、救护车、家庭等,以及可能会影响其性和有效性的环境条件(如温度、湿度、功率、压力、移动等)。 ③详述产品适用人群,包括目标患者人群的信息(如成人、儿童或新生儿),患者选择标准的信息,以及使用过程中需要监测的参数、考虑的因素。 ④详述产品禁忌症,如适用,应明确说明该器械禁止使用的疾病或情况。 ⑤已上市同类产品临床使用情况的比对说明(需列表比对)。 ⑥同类产品不良事件情况说明(需列举相关引用资料,如果有不良情况的发生,应说明如何避免)。
Q5: 一类器械备案的资料递交方式有哪些? A5: 目前一类医疗器械备案的资料递交根据不同的市局要求不一样,有些是省局网上申报,现场提交纸质版,有些是全部电子版提交。 Q6: 一类备案审批时间多长? A6: 一类备案为现场审批。也就是现场就可以拿到备案凭证。 Q7: 一类有源医疗器械备案是否要做电磁兼容测试? A7: 如果产品属于一类有源医疗器械,则需要增加安规和电磁兼容(EMC)方面的检测报告。
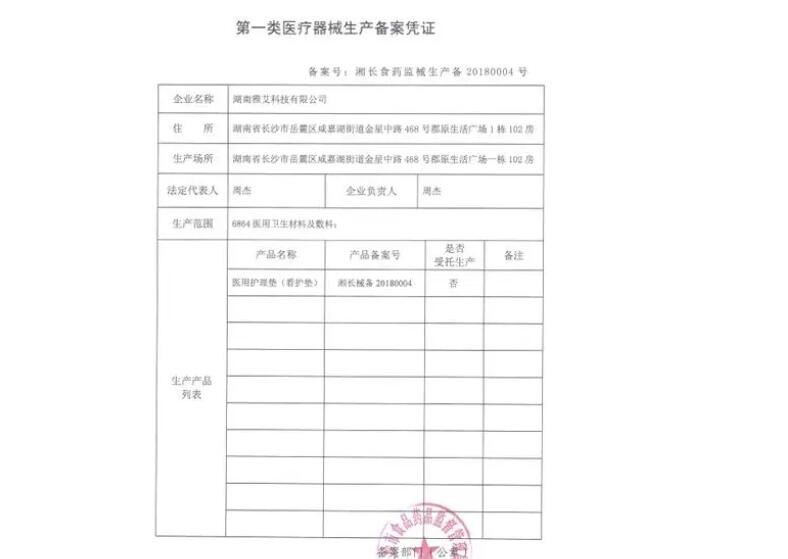
类医疗器械备案凭证包括:《类医疗器械备案凭证》和《类医疗器械生产备案凭证》。 《类医疗器械备案凭证》是该产品的批准证明,相当于产品的身份证,有了这个凭证,该产品也就有了合法的注册资质。 《类医疗器械生产备案凭证》是该产品被允许生产的批准证明,相当于企业的生产许可证,如果没有该凭证,该企业不可以生产该产品。 生产企业应该先办《类医疗器械备案凭证》,拿到了这个凭证,才能办《类医疗器械生产备案凭证》。否则《类医疗器械生产备案凭证》的信息将录不上去。这与过去先到省里办类生产登记,再到市局办理类注册的顺序正好相反。
办理一类医疗器械生产备案,企业应具备的条件 1、有与生产的医疗器械相适应的生产场地、环境条件、生产设备以及专业技术人员; 2、有对生产的医疗器械进行质量检验的机构或者专职检验人员以及检验设备; 3、有保证医疗器械质量的管理制度; 4、有与生产的医疗器械相适应的售后服务能力; 5、产品研制、生产工艺文件规定的要求。
- 公司: 河北奇源企业管理咨询有限公司
- 主营: 河北一类医疗器械生产备案,河北医疗器械网络销售备案
- 地址: 河北省石家庄市长安区胜利北大街289号财富天下一号楼二单元804
- 联系: 张经理
- 手机: 19536943631
-
微信: